
Twelve drugs have done more than $10 billion in annual sales, and seven of those were biologics—a category of drugs that are 1,000× larger than aspirin, assembled out of natural amino acids that break down into basic nutrients, and grown in single-cell factories rather than synthesized with chemistry.
I'm joined this week by Brian Finrow, CEO of Lumen Bioscience, who thinks we would be better off if we grew more of these drugs in food-grade algae, and fewer in sterile-grown Chinese hamster ovary cells.
We talk about the biochemical properties that make monoclonal antibodies particularly safe and effective as drugs—but incredibly expensive to manufacture—and whether the fundamental problem of the biotech industry is that everyone just has too much money.
Quotes
"The inside of the tube is...the outside of my body" (28:44)
Brian Finrow:
This is something we think is just dead obvious: this is a disease of the GI tract. These bacteria are in the tube—the lumen—of the GI tract, and we can put the antibody drugs right there, where they are.
Now, contrast that with—if you inject an antibody against C. difficile infection, it goes in, into your arm usually—and almost all of that antibody protein stays in the blood.
Ross:
Right. This is gonna diffuse through, like, eight liters of blood, and then some of it's gonna be in my gut, I guess—
Brian Finrow:
—and in very small amounts.
Ross:
Or rather—it’s going to be on the outside of the gut…
Brian Finrow:
Scientists have a term for this, they call it the partition coefficient—for any given protein, what percentage of the concentration of the drug in the blood serum—what percentage of that concentration do you achieve in the interstitial tissues?
Estimates vary, but it’s something like between 1% and 0.1%. And remember, too, for C. diff, the disease isn't in the interstitial tissues between the blood vessel and the GI tract. It's in the tube.
Ross:
It's on the outside of my body—the inside of the tube is, topologically speaking, the outside of my body—
Brian Finrow:
(laughs) Yeah, so you have to inject the drug and hope enough of it leaks out of your body to get to where the infection is.
"It’s a seed round, you know, for $200 million." (1:05:14)
Brian Finrow:
A lot of the cost comes from complexity. And we’ve never— I saw a newspaper article in the trade press a couple weeks ago... The headline was, “Such-and-such company announces $200 million seed round.”
Ross:
(laughs) What. What?
Brian Finrow:
Right? (laughs) That’s—it’s incredible. It’s a seed round, you know, for $200 million.
Ross:
…why did they need $200 million before they’ve filed for their first-in-human?
Brian Finrow:
I mean, those funds have gotten so big. Back when I was practicing law, a typical biotech fund was $300, $400, $500 million—like the big ones—and then they would do equity financing deals that would be $5 million, $10 million for a Series A. Now that’s not even a seed round! (laughs)
Ross:
Yeah, $5 million is just a pre-seed, just to get something done, but when you’re at Series A, it’s $30 million to $50 million—that’s what people say.
Brian Finrow:
It’s incredible, yeah. But we never had that kind of money—not for a lack of trying, but we can talk about that later. [Lumen] is just not really a fit for what the VCs look for and want to do.
Ross:
Well, it doesn’t look like everything else.
Brian Finrow:
It doesn’t look like everything else.
"…at a price that’s affordable, independent of government solvency." (1:29:51)
Ross:
If we live in a world where therapeutic proteins—whatever you want—cost $10 a gram, what is the single thing you’re most excited about? For human flourishing, for health, for you and your kids…
Brian Finrow:
Our costs at scale are already well below $10 per gram equivalent pure protein.
And at those levels, here’s an idea—I find it discouraging to think about the reimbursement system, how drugs are purchased, distributed, allocated—the policy choices about the allocation made by insurance company formulary managers—
Ross:
—in smoky back rooms, away from—
Brian Finrow:
—and by government agencies, also without a ton of scrutiny. Not just in the US, also in Europe and other places.
So I like thinking about the opportunities for some of our products. Our weight loss product is a great example because it’s already a hybrid market in the US—
Ross:
—as that market is going direct-to-consumer, as well as through the old [insurance] system.
Brian Finrow:
Up until now, it’s been so expensive to make biologic drugs that the only people who can afford them are the ones with rich governments. It’d be nice, if we can get the cost out of it, to sell those at a larger scale—still do a good job for our shareholders who took a lot of risk backing us—but sell it at a price that’s affordable, independent of government solvency, and thereby unlock a much larger market.
That’s an exciting vision for the future. I don’t think many companies are working on it, but I think people are heading in this direction right now.
Timestamps
1:17 - What makes a molecule a drug?
4:48 - What’s so great about antibody drugs?
5:59 - The humble Chinese Hamster Ovary cell
11:27 - Introducing: Lumen Biosciences
12:40 - Why spirulina?
19:14 - Why isn't everyone doing this?
23:53 - Going after gut infections
33:44 - “Cocktails” of protein drugs
50:25 - Other diseases of outside-of-body lumens
1:04:36 - How to run a different breed of biotech
1:17:44 - “Everyone just has too much money”
1:21:07 - Extinction events…and “little mammals scurrying around”
1:24:28 - Wild speculations about the future
Transcript
Introduction (1:00)
Ross:
Welcome to Development & Research, where we talk to people doing things differently in the world of clinical trials and drug development. My guest today is Brian Finrow, who runs a company trying to make drugs cheaper by growing them in giant vats of algae.
...do I have that right?
Brian Finrow:
That’s right, yeah.
Ross:
(laughs) Welcome, Brian.
Brian Finrow:
Thanks for having me.
What makes a molecule a drug? (1:17)
Ross:
I want to start at the level of a single molecule, and we’ll work our way up to the company and a medical system. So this might be a basic question, but what makes a molecule a “drug”?
Brian Finrow:
(laughs) That is an ontological question...and it’s more complicated than you’d think.
Strictly speaking, a drug—in the FDA’s rules and statutes—is anything intended to treat, prevent, or diagnose a disease.
Ross:
Sure, but when I look at the physical reality of something we think of as a drug, it’s a molecule.
Brian Finrow:
Yes. And at a fundamental level, if you say, “Look at the molecule, what characteristics does it have?”, we basically have three kinds of drugs in the world today. I’ll set aside the most-famous, vaccine molecules, and talk about the other two: small-molecule drugs and biologic drugs (which are large macromolecules).
What differentiates the two—of course the FDA is interested in many different characteristics that give them different ‘personalities’—but the main difference is simply the size.
Ross:
But the most important part of the molecule is the shape that the molecule has?
Brian Finrow:
That’s right.
[Ross: What do I mean by “shape”? Well, nearly every biological process is run by proteins that have just the right physical shape—plus placement of electrical charges on its different parts—to act on other proteins that act on other proteins… And nearly every drug works because it has one end that sticks to an important part of a protein, either to block its “active site” and decrease its activity, or to pull it into a more-active shape and increase its activity.
So the main thing that matters about a drug—small-molecule or biologic—is that it has one part that’s the right shape, with the right electrical charges in the right places, to stick to some biologically-relevant protein. It's just barely more complicated than saying “the thing that matters about a key is its shape”. And you can get to a drug molecule of the right shape by designing the placement of every individual atom, or by designing a huge protein with just one facet that’s the right shape.]
Ross:
The penicillin molecule is just a weirdly shaped cluster of carbons, nitrogens, oxygens made by a fungus to fight bacteria. Whereas, when we’re making a biologic drug, it’s probably a protein made by sticking amino acids together. These are big blocks, like we’re using LEGOs instead of injection molding plastic into exactly the shape we want. So now we have to deal with what the shape of a LEGO is—though, if we're building a sufficiently big structure, I guess that’s fine.
Brian Finrow:
Exactly right, yeah. Maybe just for a sense of scale, if this paperweight here were aspirin, then Humira, the best-selling antibody drug in the world (Brian: actually recently overtaken by Keytruda, but they're the same size) would be about the size of this entire building. So much, much larger, just entirely on a different scale.
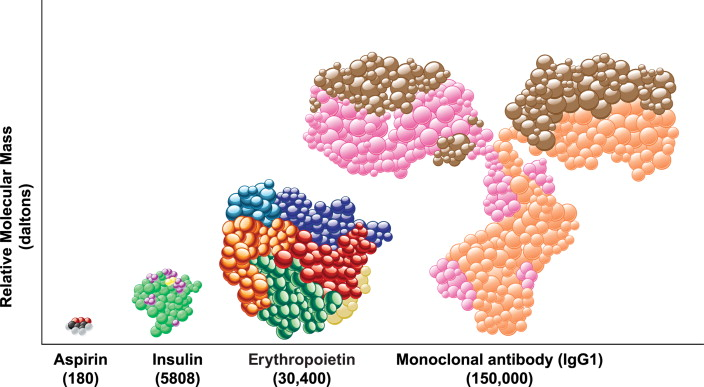
Brian Finrow:
This size difference has important implications for manufacturing. In most cases, you can synthesize small-molecule drugs with basic high school chemistry stuff: test tubes, mixing things together, presence or absence of a catalyst, heat them up, cool them down—and those processes tend to be extremely scalable. That's why drugs like aspirin are very cheap—particularly after they go generic—and are widely available. Aspirin's one example; antibiotics are another.
Now, biologic drugs—by contrast—are so complicated that we can't use those kinds of chemistry approaches. The only way to manufacture those is in some kind of a living cell. We have to hijack the existing machinery that nature gave us, through genetic engineering, to coax—in the case of Humira—it's a mammalian cell called a Chinese hamster ovary cell, into doing something it wouldn't normally do, which is to pump out a whole bunch of this monoclonal antibody protein that is the Humira drug.
(skip ahead for more about the challenges of growing CHO cells)
What's so great about antibody drugs? (4:48)
Brian Finrow:
What makes antibodies great for this [Ross: specifically, Humira blocks a receptor that causes inflammation in patients with arthritis; Keytruda blocks a key defense mechanism shared by many types of cancers.] is the biochemical properties that we call affinity and avidity, which just mean how hard they stick to their target. But they also have this property of specificity, which means they only interact with the targets they're directed at, by and large. Antibodies are famous for this specificity property, and it means that they're extremely unlikely to interact with anything else in your body and cause off-target effects.
Ross:
It's not gonna get stuck to the surface protein of one of my cells that needs to pump something—
Brian Finrow:
—exactly, yeah. So that means that, as a drug, it decreases the amount of side effects and risks that you would expect to encounter in clinical use. So antibodies are great for applications like these.
Ross:
They also get treated differently by the liver—they get metabolized differently?
Brian Finrow:
If you inject them, you know, antibodies are just proteins, and all proteins are just made of amino acids, like the rest of your body. And so when they break down, they just break down into amino acids, which are nutrients—
Ross:
—and not, like, toxic half molecules that can get in my liver and do bad things there.
[Ross: As I understand it, the reason that antibodies are more sticky and more specific—as compared to small-molecule drugs—is mostly because they have roughly twice the surface area on the “sticky” part (together with the ability to modify the sticky end into billions of different variant forms). To use our key analogy, it’s a key that’s twice as long, so it’s far less likely to bind to something undesired by mistake.
Add this to the advantages in how they break down in the body—as Brian explains—and that you can try thousands of designs in parallel through a mutate-and-check process called biopanning, and you get a pretty attractive general-purpose platform for making drugs (at least within the dominant stick-to-a-protein-target paradigm).]
The Humble Chinese Hamster Ovary cell (5:59)
Ross:
So we have this very specific sort of cell culture that someone originally captured, made tweaks—I mean, it’s a marvel of modern engineering, getting it to do this unnatural thing, pumping out this particular antibody out of these Chinese hamster ovary cells, which is not what they’re used to doing.
But…why is it that hard? I think of golden rice, which was genetically modified to produce vitamin A—so much so that its color is modified from white to gold. That modified crop can help entire countries with vitamin A deficiencies in their populations without anyone having to do anything in a factory.
These Chinese hamster ovary cells are grown in very, very controlled conditions—but if you want to deliver vitamin A, you can just hack it into rice and grow it by the acre. What’s causing the jump from “just put vitamin A in the rice and put the rice in a field” to “we have to grow Chinese hamster ovary cells in these intense lab conditions?”
Brian Finrow:
Well, I can answer that from a couple of different angles.
One is, one does not simply genetically engineer rice. It's actually quite difficult to engineer a plant cell.

Ross:
It’s another marvel of the modern age!
Brian Finrow:
Exactly. Even in the era of CRISPR—though this isn't a field that my company does work in—my understanding is that it costs an estimated $100 million to build a stable, scalable, genetically engineered strain of something like rice. It's quite difficult.
Now what makes it very scalable to manufacture once you have done that is the fact that we've got maybe 8,000 or 10,000 years of human ingenuity into making that a scalable system, in terms of domesticating rice. We haven't done that for 10,000 years with Chinese hamster ovary cells.
Ross:
Maybe we’ll be better at it 10,000 years from now! Whereas now, the growing conditions here are very intensive laboratory conditions. Can you paint us a picture of that?
Brian Finrow:
It's the opposite of planting a bunch of rice out in a field. These hamster ovary cells, I mean, really, if they had their druthers, I think they'd prefer to grow inside a Chinese hamster's ovary.
But instead, we have to trick them into growing in a giant steel tank—and that’s not so easy. In addition, the hamster ovary cells, they're evolved in that particular setting to make the proteins that a Chinese hamster ovary needs, not human monoclonal antibody. So we also have to trick it into that second trick.
All of that requires an immense amount of effort, and the tricks involved really explain why it is that over 100 years elapsed between when—in the late 1800s—Paul Ehrlich first coined the phrase “magic bullets” to describe antibody drugs and the 1980s—when they got the first antibody drug into human clinical trials. So that's all part of the manufacturing side. But on the other end of the spectrum, there's also the domain in which it's being used.
[Ross: A bit of history on “magic bullet” drugs—in the late 1800s and early 1900s, the only anti-infective chemicals available were toxic to humans as well as bacteria, and good only for external use. Paul Ehrlich, who would later win the Nobel Prize, postulated that, in order to kill microbes, "wir müssen chemisch zielen lernen" ("we have to learn how to aim chemically").
Ehrlich’s original work was on antibodies that could bind specifically to bacterial cell features, though his first magic-bullet breakthroughs would come from experiments with small-molecule chemicals like arsphenamine, mostly derived from synthetic dyes, which were a hot area for research in the field of chemistry. As Brian says, it would be another 80-90 years before anyone succeeded at making "magic bullets" out of antibodies.]
So golden rice—with vitamin A in it—well, you're gonna eat that, aren't you? Eating a protein, even a genetically engineered one—the default setting on that is “safe”. We eat all kinds of things every day, there’s all kinds of contaminants in all of our food, and our digestive tracts have evolved to handle that.
But if you’re going to inject these foreign bodies—because that’s what drugs are—then you have to be extremely careful, because there are so many ways to harm someone with the wrong thing. For one concrete example, there are mammalian viruses that infect the hamster cells and then infect humans—you have to be very careful that those don’t make it into the vial—
Ross:
—especially if you're injecting them straight. If you were eating them, you'd have some lines of defense with acid and the immune system and all that.
Brian Finrow:
Exactly. You can also get ground glass. You can have accidental mix-ups. The proteins are not just dry powders, right? They're mixed into a solution. They're stored in—
Ross:
—and there’ll be a whole bunch of other things, other proteins, lipids and whatever that the ovary cells are making.
Brian Finrow:
Oh, you have to be very careful to separate that out. Particularly—insulin isn't made in Chinese hamster ovary cells; it’s made in E. coli., and E. coli manufactures this other molecule called LPS, lipopolysaccharides. These things are extremely immunogenic if you inject them. People get very sick if you're not careful about getting it all out. [Ross: Wikipedia tells me that LPS is another name for endotoxin, which we’ll encounter later this season as well.]
So yeah, it's both the difficulty of making it and the difficulty of—once you've made it—making it safe to inject. All of that adds up to cost and difficulty and challenges in the drug development process as well as the drug manufacturing process.
Introducing: Lumen Biosciences (11:27)
Ross:
That brings us to Lumen Bio, your company. (That’s Lu-MEN, not Lu-MON, for all of our Severance watchers out there!)
Lumen has a particular approach to this where you’re doing something that’s more like growing drugs in golden rice, rather than hamster ovary cells in steel vats.
Brian Finrow:
Yes. We manufacture biologic drugs—just like Amgen does with their Chinese hamster ovary cells—these are still big, “building-sized” molecules, not the ones the size of this paperweight—
Ross:
—and they’re being built by living things?
Brian Finrow:
That’s right, they’re also being built by living things. But there are two key differences:
- First, we’re not making these for injection. All of our current programs in the clinic are for disease targets in the GI tract and GI tissues with oral delivery.
- Second, instead of using Chinese hamster ovary cells or E. coli cells—which are considered the workhorses of the biotechnology industry—we work with spirulina, a different microbe which is itself a food.
Ross:
The shop down the street sells spirulina smoothies with kale and spinach—I’ve actually got one here. You’re growing drugs in that same stuff?
Brian:
That's it. Exactly the same stuff. (laughs)
Why spirulina? (12:40)
Ross:
Alright, why spirulina? Why not rice? I think we understand why not Chinese hamster ovary cells [if we can avoid it]. But why spirulina specifically?
Brian:
So, people have tried this idea in rice. A Japanese pharma company, Astellas, has done a lot of work making therapeutic proteins—and even vaccines—in rice seeds. Others have tried with tobacco leaves, corn, potatoes—but there are some very special things about spirulina that make it particularly useful for this kind of application.
If you’re growing your drug in rice seeds, for example, you are going to grow a lot of plant material you don’t use. Most of a rice plant is not, in fact, rice. You’re going to have to grow entire fields of it, then cut it down, and throw most of it away. There’s an efficiency element there. Spirulina, by contrast, is a single-celled organism—a blue-green algae—grown in saltwater, and that’s very simple.
It’s got another advantage—a little more technical. If you think about what’s in most foods like potatoes or rice or soybeans, it’s mostly carbohydrate or lipids—because the plants are using these carbs to store energy. Spirulina, by contrast, is about 70 percent protein—
Ross:
—which is why people keep trying to put it in my smoothie.
Brian:
Exactly. That’s also because there’s a lot of other vitamins in it, but the protein content is also why it’s useful for us, because the drugs we’re making are proteins. So if you think of these things as miniature protein-making machines—they just love cranking out protein—
Ross:
—then there's a lot of protein, so if I hijack a quarter of the protein, I'm gonna get a lot of protein. If I hijack a quarter of the protein in a, you know, rice plant, I'm gonna get a tiny bit of protein.
Brian Finrow:
Yep. This really matters in terms of cost, of course, because the more protein you get to express in your cell (or whatever your host organism), then the cheaper it is. You're not wasting resources growing other parts of the plant that don't have utility.
It's also important because—remember—we need to get this into a capsule. With our best performing lab strains of spirulina we've gotten as high as 20% to 25% of dry biomass as soluble bioactive protein, which is hugely important. The highest I think I've ever seen reported in a rice seed—and they had to go to extreme lengths to achieve that—was maybe 0.1%, so dramatically lower.
Ross:
You haven’t improved it by 25 times, you've improved it by 250 times?
Brian Finrow:
Yeah, [spirulina] is just much better at this particular task than rice. So even though rice is cheaper at the grocery store than spirulina, that’s a big advantage for this application.
Now the spirulina in your smoothie almost certainly comes out of the Imperial Valley in California. They have those huge outdoor ponds of 100 acres or so. They grow it there, which is another clue—spirulina is the only microbe that is farmed at such a scale.
Any other microbes we're using, in fact, all of the three workhorses in biopharma—E. coli and yeast and Chinese hamster ovary cells—they have to do everything in sterile tanks. But think about it this way—they can make that cheap enough to sell in the grocery store, which is a clue to the scale they must be growing it at, because economies of scale are the flip side of scale.
Ross:
And they're either filtering out the E. coli that makes it into the pond or the E. coli never makes it into—well, they're big open air ponds, the E. coli is gonna drift in—
Brian Finrow:
There's all kinds of stuff in there. In fact, this particular one is on a migratory bird route.
Ross:
Nice! So some particularly biological things are going to be in it…
Brian Finrow:
Then a lot of fly larva comes out of it in the summer. You don't always want to think too much about what's on your food.
Ross:
(laughs)
Brian Finrow:
It's not extremely clean. It's just actually a testament to how well-evolved our GI tracts are that we're not all sick constantly.
Ross:
So your spirulina vats are cleaner than the big open air ponds on those migratory—
Brian Finrow:
Yes, so this is a point I want to emphasize: we’re not growing it in the pond with the birds. But it is remarkable that even with that production system it’s still totally safe.
Now of course we’re making biopharmaceuticals—some of these go into people who are very sick, so they’re very vulnerable. We grow indoors, where we control all the inputs and the environment—but the key thing is it's not necessary for it to be sterile in our system. It’s indoors and much, much cleaner but sterility isn't required and that's a key to getting the cost down and the scale up.
Ross:
So rather than a stainless steel vessel—and then we have to sanitize it afterwards and maybe we failed and a little microbe comes in [and ruins a whole batch]. Instead, your spirulina vats—this is technology that beer brewers are using—
Brian Finrow:
Our equipment is even cheaper than what they use to brew beer.
Ross:
Excellent. (laughs)
Brian Finrow:
And the capital expenditure, the cost of buying all that gleaming stainless steel and all that piping—particularly on that downstream side—if you break down the cost of one dose, out of a biomanufacturing process, the [amortized] capital expenditure to build the facility and equipment is typically 60–70% of your cost per dose.
Our goal was to get the cost per gram of protein therapeutic equivalent down by at least one hundredfold, and you cannot do that unless your capital expenditures are dramatically lower than on a traditional system.
Why isn't everyone doing this? (19:14)
Ross:
So I have been to your factory floor, and you’re set up in a former bakery in Seattle. You’re a pre-revenue company, and you have not sunk all of that capital cost in—that’s all because of the organism that you have chosen to grow your drugs in? That’s what’s been enabling that and making it possible?
Brian Finrow:
That’s right.
Ross:
…why isn't everyone doing this?
Brian Finrow:
Well, everyone tried. (laughs)
We have some very nice IP around the technology necessary to bio-engineer the [spirulina] cells to make a therapeutic drug protein. My co-founder Jim Roberts and one of my former colleagues, Ryo Takeuchi—the two inventors on the patent—made a real breakthrough. You see, spirulina's been on the market in the US since the '70s—
Ross:
—and [indigenous Americans] have farmed it and eaten it for centuries—
Brian Finrow:
—and people have been making recombinant protein therapeutics since the 1970s, too. [Ross: “Recombinant” DNA is artificially created DNA that combines genetic material from different sources. “Recombinant protein” means protein produced by adding DNA for making it to a “workhorse” cell.]
So this is kind of an obvious idea: why don't we use spirulina to make [recombinant protein]—you know, put the two together? And many very well-funded groups tried and failed over the years. Occasionally, they do us a favor; they publish their failed attempts—which scientists usually don't do—which tells you something about how often people came back to this problem. None of them succeeded for half a century, until Jim and Ryo figured it out.
So in a way, the company we aim to emulate is Amgen, because like Amgen, we are built on a fundamental breakthrough in cell-engineering technology. For Amgen at the time [in the 1980s], the new thing on the scene was mammalian cell engineering, and that's what allowed them to make EPO and then monoclonal antibody drugs, Enbrel and all these other things. It was a real breakthrough that set the stage for sequential innovation by opening up a new field. Very similar, actually, to Genentech, which was founded—in the late 1970s—on then-new technology for engineering E. coli for the first time. So that's a grand tradition we aspire to carry on.
Ross:
Where the story is, we can manufacture something that we couldn't before—we can manufacture something in a way that we couldn't before—and now with that power, there are any number of drugs that can come after that and address [diseases] that couldn't be done before.
Those drugs—it wasn't possible to produce them at all, at the right scale, at the right cost—those drugs are only made possible by this breakthrough in manufacturing.
Brian Finrow:
Yeah, that’s exactly right.
But we're not trying to put these other platforms out of business. For example—I'll start with E. coli, in the 1970s. At that time, the very obvious thing was to make insulin. Before then, the world got its insulin from pig pancreas.
Ross:
Like, you have to grow a whole pig—and then you slaughter the pig and grind up its pancreas—
Brian Finrow:
(motions squeezing insulin out of a pig pancreas)
Ross:
—oh dear.
Brian Finrow:
Yeah. Also, we have much better insulin today! We just grow the E. coli and you don't have to waste all the time growing up the snout and everything.
But the reigning drug technology at the time was small-molecule drugs—antibiotics and things—these guys were all ruling the roost. It's not like Genentech shut down or out-competed those legacy technologies. In fact, they're still making small-molecule drugs—and there are new drugs coming out on those technologies! It's just that the new technology for E. coli allowed you to do a thing that you couldn't do with the small-molecule technology—
Ross:
—so it’s just adding to the field. (thinks) Well, it was maybe putting the pig-insulin industry out of business…
Brian Finrow:
It did put the pig-insulin people out of business, that’s true.
Ross:
...but it meant that so many more people could be helped by insulin.
Brian Finrow:
Eli Lilly is who got that IP from Genentech and they're a $700 billion company now. So I think they're doing fine.
Ross:
They've done a few things after that—especially recently—but, yeah.
[Ross: In particular, they began this decade as a $130 billion company, and have benefited tremendously from a surge in GLP-1 obesity drugs. Now, you might argue that they were in the best position to dominate that market because of their position in the diabetes-management market that began with their insulin program…but I’ll leave that speculation at that.]
Brian Finrow:
So then in the '80s, [Amgen] came up with mammalian cell engineering. And again, there are things you can do in mammalian cells you can't do in E. coli—for example, make a monoclonal antibody. So it's not like Amgen put Genentech out of business; there's just a whole new wave of innovation that came out of that.
And interestingly, it wasn't until maybe 10 years ago that for the first time, the majority of new drugs approved by the FDA were actually biologic drugs.
Ross:
Oh, but we have now gotten to that point where the majority of new drugs are made out of these big things made out of biological building blocks, built in biological processes?
Brian Finrow:
Yeah. But the E. coli people are still in business. The small-molecule people are still in business. We're just making the pie bigger and bigger.
Going after gut infections (23:53)
Brian Finrow:
And so you asked, what's the “killer app” for us? Well, let's start with the easy things. Our most advanced program is directed at a disease called C. difficile infection, which is a bacterial infection of the GI tract—so it’s great for us.
Ross:
If I get a bad bacteria in me, then it starts to colonize my gut, or—who gets it?
Brian Finrow:
So, C. difficile is a bit interesting because about 10% of people—at any given time—have a little bit in them. It's all around us in the environment. You can't avoid it, particularly if you visit hospitals.
Normally, though, it's fine. If you have a healthy immune system, if you're a mid-30s, healthy adult with a good Western diet, you're fine. It doesn't trouble you at all.
Ross:
It's just in this ecosystem of bacteria that are in my gut and they're in balance and everything is good—
Brian Finrow:
—but then, what typically happens is, it's usually an older person, they have weaker immune systems, they go to the hospital to get antibiotics—usually for something unrelated, like a hip surgery or whatever—clindamycin, in particular, is notorious for this.
So they get their clindamycin. It clears up their infection in their surgery wound—but also kills off their GI microbiome. And it turns out that's very important for keeping this C. difficile—which is all around us—at bay, as our commensal bacteria crowd it out.
But then, when your commensal bacteria are damaged by antibiotics, then that C. diff kind of moves in, sets up house, starts pumping out this toxin—TcdB it's called. That toxin breaks down the cells lining your gut and then you get all these terrible symptoms, diarrhea, culminating sometimes in fatalities, and sometimes in colonic resection surgery—meaning, you live with a colostomy bag the rest of your life. So it's truly terrible.
Brian Finrow:
So that's the disease; what's it got to do with us?
Well, remember what we're doing here is we're using this food organism that we can now engineer and grow safely with these drug proteins. And in this case, we're making these proteins—they're not strictly monoclonal antibodies, but they’re kind of like that.
And what they do is just like—during Covid, remember how we had the antibodies against the Covid spike? [Ross: See examples 1, 2, 3, 4, 5…] If the antibody binds to the COVID spike, [the virus] can't infect your cell. Antibodies are great at this. This is what your body uses, manufactures anyway, to fight off viral or bacterial infection—
Ross:
—by sticking to them. That’s what they're engineered for.
Brian Finrow:
Yeah. So what we do is we find an antibody protein that sticks to the part of the toxin that causes your gut lining cells to break down, and it neutralizes them—like an antidote to the toxin.
Ross:
Do they pass through the stomach and the acid and all of that?
Brian Finrow:
So this is a funny thing, a very serendipitous discovery—we didn't know this going into our first clinical trials. The way we make this drug product, the protein molecules are inside the [spirulina] cell. And then they're dried, so it kills the cells—they're not crawling around inside you or anything. They're dead cells, they're inert.
But nevertheless, they're intact. And so it's like a little bio-encapsulation. And it turns out in the low pH [Ross: meaning, the acidity] of the stomach it all stays together until it's released into the small intestine, which is where most GI infections happen. In the small intestine, there's a big change in the acidity—it gets much more neutral—and then the cell breaks open. It's very elegant, actually.
I wish I could say we designed it that way, but it's just a convenient coincidence.
Ross:
But this is just how my body is breaking down all different kinds of the foods that I eat—or, some kinds of plant cells—but because of something about the spirulina cell, it's mostly getting released in the GI tract. And so if you want to go into the GI tract to fight an infection with the proteins inside, you're golden.
Brian Finrow:
It’s perfect. And we've published all of this data—it's really kind of an elegant solution that makes it very inexpensive for us to make these products.
What makes it particularly well-suited—coming back to your original question about what we do that other older platforms don't do well—is something we think is just dead obvious: this is a disease of the GI tract. These bacteria are in the tube—the lumen—of the GI tract, and we can put the antibody drugs right there, where they are.
Now, contrast that with—if you inject an antibody against C. difficile infection, it goes in, into your arm usually—and almost all of that antibody protein stays in the blood.
Ross:
Right. This is gonna diffuse through, like, eight liters of blood, and then some of it's gonna be in my gut, I guess—
Brian Finrow:
—and in very small amounts.
Ross:
Or rather—it’s going to be on the outside of the gut…
Brian Finrow:
Scientists have a term for this, they call it the partition coefficient—for any given protein, what percentage of the concentration of the drug in the blood serum—what percentage of that concentration do you achieve in the interstitial tissues?
Estimates vary, but it’s something like between 1% and 0.1%. And remember, too, for C. diff, the disease isn't in the interstitial tissues between the blood vessel and the GI tract. It's in the tube.
Ross:
—which means it's on the outside of my body—the inside of the tube is, topologically speaking, the outside of my body—
Brian Finrow:
(laughs) Yeah, so you have to inject the drug and hope enough of it leaks out of your body to get to where the infection is.
Ross:
Yeah. And so you would be lucky if 1% of it leaked, and so you need to be treating it at 100× the dose when it goes into your arm…
Brian Finrow:
That is an intuitive way to understand it. Particularly for C. diff, because actually a large pharmaceutical company did design and develop a monoclonal antibody, IV-infused for C. difficile infection. [Ross: The company was Merck, but Brian is too well-coached to say the name of a competitor on-camera.] It's exactly what you said—you gotta inject enough of it.
So the dose size there was 15 milligrams per kilogram—which is a whole gram of pure antibody protein, was the dose. [Ross: That’s something like 2% of your daily recommended value of protein, by the way, in the form of these hamster-ovary-grown antibodies being infused into your blood in an IV.] There are very few monoclonal antibodies that have been approved for infectious diseases—and this is something they have in common, these huge, huge doses. The top dose in Regeneron's Covid trial—which has the same issue, because the disease is in the airway, on the outside of the body—was 15 grams. Just huge.
[Ross: By contrast, Keytruda for cancer is usually given as a 0.2mg dose and Humira for inflammation is given as a 0.04mg dose for adults.]
And because this large pharma company put so much work into this, we actually have some really nice data from that. They looked in the stool of these patients with C. diff, and they were only able to detect very minimal quantities in fewer than 10% of the subjects in the study, 10% of the samples.
Ross:
And in 90% of them—
Brian Finrow:
—they couldn’t see it at all. And [in the 10%] the amounts they could see were not even large enough to quantify, so just minimal trace amounts. So it's kind of remarkable that that drug worked, actually.
Now compare that with what we do, and we can just fill up the whole tube.
Ross:
And maybe 1% of it leaks in the other direction, I guess.
Brian Finrow:
We have no evidence of that, but it could be happening. [Ross: Spoken like a lawyer, I guess.]
Ross:
Or maybe, in 10% of patients, an unquantifiable amount leaks in the other direction.
Brian Finrow:
It could be. But in general, we get these two pretty nifty advantages for a disease like this. First, we get a better shot at efficacy because—well, pretty much every drug has an effect that is dependent on its concentration, and we can get more concentration to where it’s needed—
Ross:
—because there's some practical limit to the concentration you can give, either by the economics of manufacturing, or by the side effects that come with greater concentrations?
Brian Finrow:
And that's the second thing—the side effects are typically driven by exposure of other tissues in your body [to the drug]. And by keeping it in the GI tract, we avoid those exposures, and we cut down on the risk of off-target effects & toxicities.
So in short, particularly for a GI disease like C. diff, it's a way of making a drug that is just everything you want. It is more likely to be more effective, less likely to cause off-target toxicities and side effects, and on our platform, because of what we were talking about earlier with the [capital expenditure] advantages and the scalability of it—remember, cheap enough to buy in the grocery store—it’s also less costly to manufacture.
And as a final thing, it's much more convenient to take oral capsules than to take an IV infusion of a very expensively manufactured drug that circulates everywhere [in the body] and doesn't mostly get to where it's needed.
[Ross: Maybe you’re wondering why Brian makes such a big deal about their antibodies being directly applied versus injected. Surely that can’t be a factor that differentiates them from other biologics developers!
Well, it is enormously puzzling to me, but it does appear that all of the Covid antibody treatments that were approved—and yes, Merck’s bezlotoxumab—are given by IV infusion into the bloodstream (where the infection isn’t). Why? Beats the heck out of me. But if I had to guess, the fact that hospital nurses are used to putting drugs into IV bags probably leads to a significant industry-wide bias towards that modality, even if it makes little sense from first principles or in terms of the practical downstream reality to patients.]
“Cocktails” of protein drugs (33:44)
Ross:
How does that show up in your clinical trials? Your clinical trial for treating C. diff is going to be different from someone developing an injected antibody—and even within that, there are design choices—smart choices you can make and less-smart choices you can make.
What are you doing differently in your clinical trials based on having these advantages, or just because everyone else gets it wrong and you might as well get it right?
Brian Finrow:
I should caveat this with, I’m not a clinician—I trained as a lawyer—
Ross:
—we’ll talk about that in a minute. But I trained as a quantitative trader, so we have two non-clinicians talking about clinical trials here.
Brian Finrow:
In the case of this particular program, we are doing something that is quite novel for the industry, in that the drug product—well, I kind of lied a little bit and over-simplified what it is.
There’s actually four proteins in it, and that is something very different than the industry does. Three of them are these antibody-like proteins that bind and neutralize Toxin B, which mediates [the disease] in humans with C. difficile infection. And the fourth, it’s a different thing—it’s an enzyme, a different kind of protein—and this enzyme interferes with the cell wall of the bacterium itself—
Ross:
—similarly to an antibiotic mechanism?
Brian Finrow:
Kind of, though antibiotics—strictly speaking—are small molecules. For this enzyme protein, the important part is that it has a different mechanism [for disrupting the bacterium], rather than binding to the toxin.
Now we didn’t do this just because we want elaborate complexity in our lives. We think that this combination will be much potent than just having any one of the proteins.
Ross:
I mean, this makes sense. My question when you say, “Yeah, we just stick [antibodies] to the toxin,” was—the toxin's just gonna stick around? You’re going to leave all the bacteria there, and they’re going to keep pumping out toxin—what's the endgame here?
But it sounds like the endgame is an enzyme to go after the bacteria itself—and hopefully the immune system is regrowing and the gut ecosystem is regrowing eventually.
Brian Finrow:
Well, here we were encouraged. Remember even in the big trial I was telling you about by this other company, mostly non-detectable levels of one antibody was enough to improve disease outcomes. So we are optimistic—we've got three, and the three together are about a thousand-fold more potent in their measured neutralizing power. So we are pretty optimistic that we got something good going on there.
Ross:
What is the thinking behind having three antibodies? For treatments that went after Covid with antibodies, was it also three antibody types?
Brian Finrow:
Generally one, sometimes two—Regeneron made one with two. What we’re doing there that is quite different, is the combinatorial exercise.
Ross:
Is this because there are three different types of toxin, or that the antibodies need to work together?—
Brian Finrow:
Much more interesting than that, actually. There is a bit of diversity [in toxin structure], but that’s not actually the main reason.
What we discovered—and we have a pre-print on this working through peer review—is that if you design an antibody cocktail (that’s the FDA term for a combination of antibodies), the artful combination like this can get us a measured potency that is thousands-fold more potent than any single antibody could ever achieve.
Ross:
What’s going on there?
Brian Finrow:
It’s super interesting—it has to do with the behavior at the molecular level of an antibody binding event. So when antibodies are binding, they’re not like sticky tape. They don’t just bind and they’re glued on; that’s a covalent bond, but these are non-covalent binding events.
Ross:
Like velcro, where it’ll stick but it could get pulled off?
Brian Finrow:
They’re more like static electricity, like a balloon that sticks to your head.
Ross:
Like if I stick a paper onto the ceiling and maybe it’ll—
Brian Finrow:
—it’ll stick for a while. But with antibodies, everything moves very fast at the molecular level. The antibodies are basically—they’re coming on and off of their substrate all the time. And you can measure this. There are different tools to measure the on rate and the rate at which they come off after the binding.
All of this gets reduced down into a single figure that’s most commonly reported, which is the affinity, or in the case of a multivalent antibody, the avidity. This dynamic behavior is what’s important to understanding why it is that we can achieve such high potencies with what we call a synthetic avidity effect.
Basically there’s three—again, not antibodies, but antibody-like proteins—and if this model is the toxin, they’re binding in different spots. And they’re coming on and coming off constantly—
Ross:
—but this one is stuck on 80% of the time, and this one is stuck on 80% of the time, and—
Brian Finrow:
—and remember, each of these alone has neutralizing potential. So what you want to maximize is the time that something is neutralizing it.
Ross:
Just thinking about it, if this one has a 90% “on” rate, and this one has a 90% “on” rate, and that one has a 90% “on” rate—then one of them’s gonna be stuck on 99.9% of the time.
And so I could spend forever and a day trying to engineer an antibody which was so sticky that it stayed on 99.9% of the time, or I take three of these on three different binding sites, which are each 90%, and I get a 99.9% cocktail.
Brian Finrow:
That’s it.
Antibodies’ potency is measured in concentration terms—how much drug dose of the cocktail do we need to cause the 50% maximal inhibition of cell intoxication of a certain amount of Toxin B?
Ross:
—to make this amount of toxin half as deadly as it was?
Brian Finrow:
Yeah. So these antibody potencies are measured in concentration, so molar terms, in terms of the concentration of antibody for whatever its effect—
Ross:
—that means, how many antibody molecules I need per milliliter, times some ridiculous multiplier. [Ross: Dr. Janet Doherty of Oakland Mills High School will be relieved to hear that I do actually know this multiplier to be 6.02 × 10^23]
Brian Finrow:
Yeah. So, a typical antibody you get out of a machine learning or AI system will be a low-micromolar binder. [Ross: Meaning, it requires roughly 10^18 - 10^20 antibody molecules per milliliter to cut the toxicity in half.]
A good antibody, a clinical-grade antibody, will actually be nanomolar, a thousand times better. [Ross: 10^15 - 10^18 antibody molecules per milliliter.]
A great antibody will be picomolar. [Ross: 10^12 - 10^15 antibody molecules per milliliter.] And there are very few examples of that.
Ross:
Are the best-selling antibodies way down there?
Brian Finrow:
The best-selling antibodies are mostly in the nanomolar range. The IV-infused antibody that I was saying earlier, that [Merck] was selling—I guess it just got pulled from the market—I think that was in the low-nanomolar range.
So picomolar is great. The measured avidity of this triple cocktail with this artful combination is in the femtomolar range.
Ross:
A thousand again? Wow. [Ross: 10^9 - 10^12 antibody molecules per milliliter.]
That means that you’re getting your effects at a million times lower dose—at literally a million times lower dose than where antibodies normally are on the market by doing this combination effect.
Brian Finrow:
Yeah. So, going back to your original question, which is what’s different about how you do clinical development? Well, this is something very different.
And it’s troublesome, because each of these is in a different strain [of spirulina], which means you have to grow more [Good Manufacturing Practice] batches and blend them together. That’s a lot of complexity. But this kind of potency advantage we think is going to be important to the ultimate clinical efficacy.
Because, remember, in the real world—I mean, this is all measured in a Petri dish in the lab, right?—in the real world, there’s all kinds of other things going on. Including, for example, people don’t always take their drugs. For C. difficile infection in particular, it’s predominantly an older crowd—there’s a lot of pill burden among these individuals to begin with, and a lot of times they’re forgetful and don’t adhere to the dosing regimen.
And also it’s a very messy environment. C. difficile is characterized by an awful lot of GI distress—diarrhea and things. It’s a chaotic environment that we’re going into, and so we wanted to whack this disease as hard as we can. And so that’s why we have this complexity.
The reason I think very few other companies do this—
Ross:
—which was going to be my next question.
[Ross: Look, I think that “we’d prefer more strength rather than less strength” is a fine justification for wanting a more comprehensive treatment; I don’t really think that it needs to be justified in terms of the C. diff treatment environment being unusually messy.
On the other hand, I have been shocked by the number of drug developers I’ve talked to who consider dose-finding a matter of “good enough” and risk minimization, rather than being actually excited about going for More Dakka on efficacy where possible. Brian is one of those that speaks my language on this score (with—to his credit—an appropriate amount of nuance on the medical context).]
Brian Finrow:
Yeah. I think we’re pioneers in this regard. Look, it’s very expensive to develop a single monoclonal antibody on one of these conventional systems—
Ross:
—and if you’re proposing to do it three times, then—
Brian Finrow:
—then it’s three times as much.
And, you know, people still encounter unexpected anaphylaxis, for example, with injected proteins—that happened with a Covid program recently. So if you inject three different proteins because you want to get the cocktail advantage, then you’ve got a real risk that, in the clinical trials—
Ross:
—any one of them has a rare side effect and—
Brian Finrow:
—and then you’re toast, yeah.
Ross:
Whereas in your setting, you’re just doing a much safer thing. And yes, it might be the case that one of the three of them has a whatever [if they were injected], but they were all very safe because you were putting them on the outside of your body anyway.
Brian Finrow:
Exactly. That’s a big advantage.
Ross:
There are other companies that are running C. difficile trials that have candidates for whatever, and some of them are trying to treat straight to the GI tract. Are you fighting with them for patients? In your trials—well, cancer trials are infamously fighting each other for their patients—are you in the same fight with the other companies developing C. difficile drugs?
Brian Finrow:
Not really. There’s only so many oncology centers in the US, and so what I’ve heard—we haven’t run into this ourselves, but I’ve heard—is that sometimes you’ll find a situation where there are three or four trials going on for a single type of cancer, and the principal investigators just have to make hard choices about who goes into which trial.
We don’t have that issue—
Ross:
—because C. diff is common enough across hospitals everywhere?
Brian Finrow:
It’s just awfully prevalent. So there are a couple of other C. difficile trials recruiting right now, but none at the same centers where we are.
There’s another distinction [between us and the competition]. Remember what I was saying earlier about C. difficile infection, how it’s caused when these antibiotics kill your microbiota?
Well, the other companies are going a different way—rather than the way we’re doing, which is going after the bacterium and its toxin. What they’re doing is they say, “Let’s just fix what’s broken. We’ll accelerate the re-engraftment of the microbiome.” It’s called a fecal microbiota transplant.
Ross:
Okay…
Brian Finrow:
So you can get there two ways, from the bottom up—right? Or from the top down.
Ross:
(laughs) Neither one of those sounds very pleasant!
Brian Finrow:
—but they put it in a capsule.
Ross:
Okay, okay. I guess that makes the top down more pleasant...
Brian Finrow:
The jokers call it, of course, a crapsule.
Ross:
(laughs) Of course.
Brian Finrow:
But it’s fine; it’s just a capsule. And the insight is simply, okay, we’ve nuked your microbiome. Let’s give you some starter seed—
Ross:
—let’s put some other bacteria there. They’re the good bacteria that live in human guts—
Brian Finrow:
—yeah.
Ross:
Though it does seem kind of indirect and fussy, and there’s still toxin while you’re dealing with the thing, and what if it doesn’t take?
Brian Finrow:
And I think there’s a practical challenge that we don’t have, which is that you can’t give antibiotics together with a probiotic, you know?
Ross:
Oh, right—if I’m trying to drop in these bacteria and hope they survive, but I still need to be taking my antibiotic—
Brian Finrow:
Yeah. So it’s a bit troublesome for the patient—they have to come back in [for a second treatment]. Oftentimes, they have to do a bowel cleanse to wash out the antibiotics, or an enema application sometimes.
But the bigger, more fundamental issue is that the antibiotics aren’t perfect. And in the case of C. difficile, there’s a rising incidence of vancomycin resistance—there’s some research reports on that anyway.
Ross:
That’s the standard of care, vancomycin? More antibiotics for the problem that was caused by my antibiotics—
Brian Finrow:
—and that’s what’s so insidious about the disease, because you get treated, and then you have recurrences, which becomes a big economic loss-driver—
Ross:
—because the vancomycin is also killing the [microbiota] in the same way that created the opening in the first place.
Brian Finrow:
So, the three antibiotics that are most commonly used are: vancomycin, fidaxomicin, and metronidazole—and in the biggest, most tightly controlled study, it was run by the big pharma company from earlier, the antibiotics cure rate, primary cure rate was only 80%.
But it’s okay—that’s not the end of the world—you can switch to other antibiotics or load up on things, but still, that’s a remarkable fact. I mean, we’re accustomed to antibiotics just kind of working right off the bat—though even that’s not true in all people, and particularly immunocompromised people like the elderly.
So, what I’m winding up to, here, is—we’re hoping in our trial to also help that rate of primary cure, as well as preventing the recurrences. And the goal—and this is why we spend so much time making the cocktail as potent as possible—
Ross:
The [FMT / fecal microbiota transplant] candidates that are working through the [clinical trials process]—they are going to come after the second course of antibiotics to treat the C. diff, right? And then you’d hope that they colonize and that the good bacteria grow back faster than the bad bacteria grow back—whereas your treatment is going to hammer down the bad bacteria the whole time.
Though maybe that’s synergistic with also implanting some good bacteria—
Brian Finrow:
—exactly.
So, in our dream world—of course, what we want is for our drug to solve all of the problems in the world. But we don’t need that to be the case—
Ross:
—because if your drug is half of the golden duo that cures people—
Brian Finrow:
—right. So in our trial, we’re dosing it concomitantly with antibiotics, until people finish their course, and the goal there is to accelerate the antibiotics and get people off of them—because they have their own toxicities and side effects—and then we’ll continue dosing for another eight weeks afterward.
That takes advantage of the fact that—unlike antibiotics, which are nonspecific, and blitz everything—
Ross:
—and if you’re taking them for weeks, you’ll definitely mess up your gut microbiome.
Brian Finrow:
If you stay on them for weeks, yeah. So what we want to do during those eight weeks is, to take advantage of the specificity of these large macromolecule drugs—to just keep out the bad bugs while the commensal bacteria are re-growing.
So in our ideal world, then, we’ll find—we hope—synergy with the antibiotics. And there’s no incompatibility between our product with the FMT-based approaches, too!
Our goal as an industry, I think, should be zero CDI cases and zero CDI recurrences. And there’s a lot of excellent ways to combine these things in the future to accomplish that.
Other diseases of outside-of-body lumens (50:25)
Ross:
So [C. difficile] seems like—in some ways—just a perfect combination of a bunch of things going right. This is an infection in the gut. Somehow, the spirulina goes straight to the small intestine, which is where I wanted it to be. You’ve got these great antibodies. You’ve got three sites on the toxin that you can make use of.
Do you have other programs running? Can you give us a taste of what those are like and just the breadth of the approaches that are enabled by this manufacturing breakthrough?
Brian Finrow:
Yeah, so it’s pretty broad at this point. The common feature is it’s essentially any disease that has some kind of a nexus—I’ll just put it that way—with the GI tract.
Starting from the obvious ones—we spent a lot of time talking about C. diff, and there are other infections of the GI tract. They’re all amenable to the same approach. The very obvious one—anybody with kids has had the pleasure of enjoying norovirus infection. That’s like Covid; it’s an RNA virus, not a bacterium, but also gastrointestinal.
Ross:
But it means—your patient is going to have GI “events” for a couple of days…
Brian Finrow:
Then, if you think about the GI lumen as being our natural home, then you can also think about the respiratory lumen, which would be the upper respiratory tract—
Ross:
—another part that’s outside of the body, but enclosed.
Brian Finrow:
So an example there would be RSV, or influenza. We can put the neutralizing antibodies into your nose.
Ross:
We’ve had some recent experience with a third virus that ends up in your nose…
Brian Finrow:
So that’s a very interesting one because SARS-CoV-2—Covid-19—comes with a tropism (meaning, where does the virus attack you?) for both the nasal passages and the GI tract, actually. So that’s a twofer. We have a program running in that area with DOD funding.
Ross:
You’ve got a spray for the nose—
Brian Finrow:
—and then we can formulate it also [as a pill] for the GI.
And then there’s also the other obvious external surface, the skin. And so we have a DOD-funded program that’s preclinical still, also looking at antibiotics for infections in battlefield medicine. There’s a big concern there.
Ross:
And these are similar approaches, you’ve got an enzyme that attacks the bacterium?
Brian Finrow:
It’s an enzyme or an antibody, sometimes a mix of things.
Ross:
Where’s that enzyme coming from? Do you have an AI system that can—
Brian Finrow:
It depends on the program. We try to mine Mother Nature as much as possible, because it’s the only thing with more compute than the AI labs. (laughs)
So, for the C. difficile program, the enzyme there is derived from something called a bacteriophage. Bacteriophage are viruses, but they can’t do anything to humans—they only attack the bacterium that they’re co-evolved with.
Ross:
They’re viruses of C. difficile that don’t know how to interact with human cells.
Brian Finrow:
Right. This is an enzyme that that virus normally makes for its own reasons [Ross: namely, to break out of a C. difficile cell once it’s replicated itself enough], but it turns out if we just manufacture it recombinantly in spirulina, feed it to people, it pops the [C. diff] cells very efficiently. Kills them off.
Ross:
And you’ve got a mix of similar tricks for battlefield infections?
Brian Finrow:
Pretty similar, yeah. And now you can kind of feel the rhythm of what we’re doing here—in battlefield medicine, it’s the same thing. There really are variants there, and so we need to cover those variants, but now it’s with a cocktail of enzymes, not a cocktail of antibodies.
Then, another infection of the GI tract, traveler’s diarrhea—that was funded by the Gates Foundation originally.
Ross:
For the benefit of the travelers, or is that just a name?
Brian Finrow:
It’s a nuisance for travelers, but the Gates Foundation’s interest in this area comes because there’s an incredible amount of infant mortality in the developing world caused by the same things that cause traveler’s diarrhea.
Ross:
Right, the things that are a nuisance for first-world adults do kill kids in the third world.
Brian Finrow:
Yeah. So that’s their interest. Same basic approach: for these bacterial and viral pathogens—norovirus and rotavirus too—we identify a cocktail of antibodies that will bind and neutralize all of their virulence factors, that’s the term of art.
Okay, so these are all non-host-directed—directed at the pathogens. What they all have in common, up until now, is that these are basically swimming around in the GI tract. But there’s no reason that, with a protein, you can’t hit some target that is related to your natural biology.
So a very obvious place to go from C. difficile infection—which is great because we have all of this data about neutralizing this TcdB protein with an antibody can help people, so that makes it much easier and lower risk for us to develop things. Well, there’s another disease that’s very common in the US called inflammatory bowel disease (IBD) that’s quite similar. It’s an inflammatory disease, not an infectious disease. But, similarly to C. diff, there’s an enormous amount of clinical data telling us what kinds of things to neutralize. In that case, it’s not Toxin B of C. diff—
Ross:
—but it’s body systems going out of whack? Where you need to push them back down—
Brian Finrow:
—it’s these immune protein signaling cells called cytokines. They’re the obvious ones—and they’re made by the body, so IBD is an out-of-control immune reaction that happens in your GI tract, outside of the context of an infection where you want the immune response. This is where it’s gone a little bit haywire. There’s lots of autoimmune diseases—rheumatoid arthritis is a common one. So this is a very common—
Ross:
—and the complications of COVID are the escalation of the immune system, mostly.
Brian Finrow:
Yeah, exactly. So here what we’re trying to do is turn down the immune dial on the immune system that’s causing it to go haywire and overreact when there’s no infection happening.
And so there, the cytokines neutralized, the very obvious ones—without giving too much away, we haven’t launched this program into the clinic yet—but the very obvious ones are this cytokine called TNF, tumor necrosis factor, and another one called interleukin 23. So again, takes advantage of some of the same principles here.
We know Humira, for example, is a TNF-neutralizing antibody. It was the best-selling drug in the world up until Keytruda. So we know that that helps IBD patients, and [a monoclonal antibody that blocks] IL-23 is also marketed under the name Stelara. And in fact, we even know because Janssen ran a combinatorial trial, just two antibodies, that together they get an even better response than either of them alone. So that gives us a lot of optimism for that program.
Brian Finrow:
And there are other programs like that. But if you step back, again, zoom out one more level, you say, “Well, okay, so we can maybe address these targets not only within the GI lumen, but also in the GI tissues. And so what other targets are there in GI tissues?” And it turns out the GI tract is a pretty interesting interface of signaling and perception for all kinds of bodily systems.
So for example, the immune system—if you talk to immunologists, a lot of times they’ll actually just come right out and say it. It’s like, “Oh, well that obviously makes sense, Brian, because the GI tract is the largest immune organ in the body.” There’s so many subsystems within the immune system interwoven into your GI tissues.
Ross:
There are immune systems—there are white blood cells there—as part of the lines of defense?
Brian Finrow:
Exactly. And they’re all constantly monitoring what’s going on in the GI tract.
Ross:
This is the defensive line of the human body, for the football fans—
Brian Finrow:
—but it’s also a sensory line.
So very far afield from anything we’re doing, there are lines of research going on right now into the importance of the microbiome for all kinds of things. And there are some papers that, for example, have found interesting effects—seeing differences between the microbiome composition of people who respond to checkpoint immunotherapy and those that don’t.
Ross:
You mean, cancer treatment?
[Ross: Checkpoint inhibitor therapy is a kind of cancer treatment that targets “immune checkpoints”—immune-system regulatory mechanisms meant to prevent your body from attacking itself, that cancer is well-known to co-opt for its own protection. The “checkpoint therapy” is meant to turn off the cancer’s “checkpoint” flags and get your immune system fighting the cancer in the right way.]
Brian Finrow:
Yeah. So it’s totally different ’cause you could say, “that cancer is not GI cancer.” But nevertheless, we see there’s something interesting going on. And the conjecture is that, well, this is because it’s the largest immune organ, and people are thinking: “Well, gee, if we can modify the microbiome in such a way that we can cause more people to be responders to checkpoint therapy, then that would be huge.”
This is not an area where we’re working in, ourselves, but I’m just mentioning it as an example of the unexplored biology that is now unlocked by the Lumen technology. Because now we can maybe start to, with precision, interrogate these things. Until now, your choice was either nuke the whole microbiome and hope for the best, or intervene in pretty clumsy ways.
Most probiotics really are almost entirely lactobacillus, you know, because it’s the easy one to make into a probiotic. But it’s just yogurt, so this can’t be right—we would’ve known by now if it was the miracle cure for everything, right? It’s probably more complicated, and there are trillions of bacteria down in there.
Brian Finrow:
So that’s one example. There’s the immune system, the nervous system, or the vagal afferent nerves, all tied up and bound up in the GI tissues—sensing what’s going on, reporting to your brain in ways that we don’t really understand.
Ross:
So you could be looking at neurological disorders?
Brian Finrow:
That’s what people have proposed. And even some chronic diseases are proposed to have gut-brain axis origins. Parkinson’s disease, for example, is one that you see written about. Again, we’re not doing any work in this area, but—
Ross:
—but it’s exciting that this is happening elsewhere in gut biology.
Brian Finrow:
And then there’s the endocrine system. The endocrine system is also all bound up in this, and that makes very direct sense, right? Because of nutrient sensation and satiety—
Ross:
—and there has been some recent interest in the field of manipulating the gut for satiety…
Brian Finrow:
A little bit, yeah. A lot of people don’t know this, the famous GLP-1 drugs—the GLP-1 in your body comes from intestinal L cells, the cells lining the GI tract—
Ross:
—and if someone has a molecule that just interacts with it in the right way, well, that seems like a story that we’ve heard before, on this episode. So you have work going on there?
Brian Finrow:
So we do have a preclinical program going in the weight-loss space. Can’t say too much about it, but it’s very encouraging.
So all of these are just kind of examples of the things we can do. But there’s one more example which we didn’t start working on ourselves directly. A Japanese pharmaceutical company approached us with this idea—and it’s a pretty good idea—that diet has an impact on a lot of things in our lives, and a surprising one is kidney stones.
So again, kidney stones are not a GI disease; it’s pretty far from the GI tissues. I haven’t heard anybody describe the “kidney-gut axis” like the way the “gut-brain axis” has a lot of followers. And nevertheless, about 90% of the kidney stones are comprised of something called calcium oxalate—these are crystals, actually. And there’s an orphan indication called primary hyperoxaluria, where there’s a genetic disorder that interferes with the body’s ability to break down oxalate. The oxalate accumulates in the blood serum, then accumulates in the kidneys, and it becomes supersaturated, mixes with calcium, and creates these crystals.
Ross:
…I’m pretty sure I don’t want crystals in my kidney!
Brian Finrow:
And when one of those breaks loose—yeah, it’s incredibly painful.
But actually most [kidney stones] don’t come from this genetic abnormality; there are other things going on, and they have gut malabsorption syndrome. People absorb too much oxalate from the diet. So it’s even harder to avoid than gluten for celiac disease patients. So one simple approach—that this Japanese pharma company approached us with, they said—“Why don’t you just make some enzymes that degrade oxalate before it can be absorbed?”
Ross:
—in the gut.
Brian Finrow:
In the gut, yes. We should cut it off at the pass, as it were. That’s another great example.
In short—everybody we talk to has new ideas. A lot of them are really… not all of them are good, but a lot of them are really good! And the sense I get is that this—why, this must be what it felt like to be at Amgen in the early ’80s—
Ross:
—when you can do engineering for the first time, when you can manufacture biologics for the first—
Brian Finrow:
It’s not like all of those things were great ideas, but look, some of them were amazing!
Enbrel—the patent on Enbrel was filed in 1989, and it’s still a bestseller. So we’re very excited about the scope of the opportunity but also the tremendous unmet medical need that is left lingering out there, despite the fact that the newest of our three big reigning champion drug development technology platforms—the injection biologics—the newest of them is 50 years old now. And there’s still a lot of human disease.
And it seems like, wow, we’ve got this new tool, allows us to go after some of these old hard problems in a totally new way. I think we’re going to find, like Amgen did in the ’80s, there’s some really great applications somewhere in there.
How to run a different breed of biotech (1:04:36)
Ross:
So this feels tremendously exciting—you’ve described, by my count, six different drug programs at different stages of things. Most biotech startups, before they’ve gotten their first drug to market—they’ve raised a monster series A round so they can get one drug through this next stage. Then they raise a monster series B round and get it through. Then they raise a monster series C round. It’s just dilute, dilute, dilute to get just one drug passing through.
So what is happening here, that you’re running so many programs in parallel? What’s making that possible? Is it just the economics of how cheap any one of them has gotten, you can be doing six at the same time more leanly than anyone else?
Brian Finrow:
It’s partly that. A lot of the cost comes from complexity. And we’ve never—I saw a newspaper article in the trade press a couple weeks ago—I don’t remember the name of the company, but the title just blew my mind. The headline was, “Such-and-such company announces $200 million seed round.”
Ross:
(laughs) What. What?
Brian Finrow:
Right? (laughs) That’s—it’s incredible. It’s a seed round, you know, for $200 million.
Ross:
…why did they need $200 million before they’ve filed for their first-in-human?
Brian Finrow:
I mean, those funds have gotten so big. Back when I was practicing law, a typical biotech fund was $300, $400, $500 million—like the big ones—and then they would do equity financing deals that would be $5 million, $10 million for a Series A. Now that’s not even a seed round! (laughs)
Ross:
Yeah, $5 million is just a pre-seed, just to get something done, but when you’re at Series A, it’s $30 million to $50 million—that’s what people say.
Brian Finrow:
It’s incredible, yeah. But we never had that kind of money—not for a lack of trying, but we can talk about that later. [Lumen] is just not really a fit for what the VCs look for and want to do.
Ross:
Well, it doesn’t look like everything else.
Brian Finrow:
It doesn’t look like everything else.
Ross:
And when you have $500 million and you’re deploying it $50 million at a time—or whatever the heck it is—they’re mostly looking for things that look like the last big success.
Brian Finrow:
Yeah, for most biotechs, there’s an idea—maybe like a “hit”, a non-optimized drug candidate from a university—and then it’s almost always something that uses an existing technology. So maybe it’s a monoclonal antibody that there’s lots of suppliers in the ecosystem that can work with that and manufacture it for you—and these things all look the same to my outsider eyes.
It’s not to say that other people aren’t doing interesting things. I mean, there’s an enormous amount of diversity, particularly like Arch and Flagship and Atlas—Bruce Booth’s outfit in Boston—I think they’re doing tremendously innovative things.
Ross:
—and we’ve got new modalities breaking through; people are using mRNA for the first time—
Brian Finrow:
—for vaccines, and then you’ve got the cell therapies and the gene therapies.
So, all of that is true, and yet—if you look and count noses, almost all of the companies are small molecules or monoclonal antibodies. There are almost no vaccine companies, aside from the Covid era. So we never had—
Ross:
—and most of those companies are raising $10 million and $100 million at a time.
Brian Finrow:
Exactly. But we never had that kind of funding and so we couldn’t execute that kind of business plan.
Ross:
…so what did you do instead?
Brian Finrow:
Where we started instead is we had the core technology—the cell engineering technology—and rather than saying, “Here’s our drug for this disease, we need some money to go do the trials,” we said, “Well, here’s a type of cell we can engineer. What might that be useful for?” (laughs) And it’s a hard question—put that way—to answer, because it actually is useful for many things.
Human disease, by the way, is only one of the categories. There’s industrial biotechnology, there’s vet pharma, there’s foods and food additives. You name it, there’s all kinds of things. And all of them come down to biomanufacturing using a massively scalable microbe like we’ve got, so it is useful for all of those things. We can’t do all of those things. We had to figure out—what’s the killer app, where should we start? What’s the number-one best thing to do, like Genentech did with insulin?
It was our good fortune that we’re here in Seattle and some folks from the Gates Foundation wandered across, and they told us all about these poor kids in the developing world dying of these infant diarrheal diseases—which used to be a driver of high infant mortality in the US too, it’s just that we figured out other ways of solving that problem—and that sort of cued us in on the opportunities in the GI tract, which otherwise is a bit of an afterthought. There are no GI health companies with $200 million seed rounds. (laughs) It’s just not a thing!
So we’re kind of rotated 90 degrees in a way that isn’t appealing to VCs. And so we said to the folks from the Gates Foundation—like we say to everybody that has a great idea—“Got any money?”
Ross:
(laughs) They have some money!
Brian Finrow:
They do. And so they actually funded some of the first development work on this, and it was our good fortune—a lot of sweat and tears goes into this too—but things worked really well right off the bat. It’s a great idea. And that’s what set us down on this path.
But we started out on this path, and the Gates Foundation doesn’t write $200 million seed round checks. It’s small money—"real money where I come from", as the saying goes—but not this kind of thing. And so we’ve always had to be parsimonious with our spending. It just happens to be our good fortune that that is what our technology platform excels at—
Ross:
—economy, efficiency—
Brian Finrow:
—efficiency, yeah. Going back to our origin story: we’ve got this way to make oral protein drugs, but we want it to be a couple orders of magnitude cheaper than a conventional sterile fermentation system.
You can get 10% out of the cost of a system via just continuous improvement—and they’ve been doing that, it’s much cheaper now to work with those systems. But if you want to take 2 orders of magnitude out, you can't just do the same thing in a more efficient way; you have to wildly rethink things—and in particular, you have to think about things with a relentless focus on simplicity and robustness. So, of necessity, that is where our organizational DNA has evolved.
Ross:
As a company, not just in your science?
Brian Finrow:
Exactly. And it’s…everything. It started on the manufacturing side of it, but now it’s how we design clinical trials—it’s how we think about developing preclinical data packages—it’s how we do the protein engineering. I mean, you name it, everything is focused on high throughput and low cost.
I can’t speak for other companies, but my sense is that when you’ve got— I was at a very well-funded company before this, and when you’ve got that much money, it changes how you approach problem-solving. A lot of problems you try to solve with money.
Ross:
You can spend your money on outsourcing—I talk to other companies—a lot of their complexity comes from, “well, we have all this money, we’ll outsource this job, this trial, this manufacturing—”
But that’s not how you’re going about things?
Brian Finrow:
If you think about doing something yourself versus sending it to a supplier—you can look up what the typical gross margins are for these companies: it’s about 50%! And that tells you—if you run the math backwards—that’s twice the price. You hope that you get, in exchange for that premium pricing, better quality, better speed—
Ross:
—but that’s if you’re doing what they’re already ready to do. If you’re doing something different, you will get charged even worse.
Brian Finrow:
Yeah. But the insight is even more fundamental: if you have a lot of money in the bank, that ends up being the lever you pull first. But almost all problems in life can’t be solved with money! It’s as true in business as it is in family life—most things aren’t money problems. So we had the blessing and curse of never being tempted with that. But also—the technology itself just sort of naturally lends itself to this way of thinking.
Now we try to go out of our way to optimize for it in a more intentional way. That is the best I can do as an explanation as to how we have so many programs and still a pretty small team—just under a hundred people. But with that hundred people, we’re doing actually many more activities than many larger companies with more outsourcing do. For example, we have our own GMP manufacturing facility that we own and operate exclusively for ourselves—actually two GMP facilities now.
We also do all of our own hit-to-lead discovery, protein engineering, cell line engineering, all of our own regulatory work in-house directly with our own people. Really, the only segment of a fully integrated biopharmaceutical company we don’t have is sales and marketing—
Ross:
—well, you don’t have a product yet.
Brian Finrow:
Yeah. But now, when we ease into a new area, this is the lens we bring, which is, “Okay, this feels like we could spend any amount of money on this, but how are we going to do it for two or three log cheaper than the standard thing?” And it always comes down to—without compromising patient safety or scientific rigor; we don’t cut corners there—but, what are the inessential bits? And there’s a lot of inessential bits.
And it’s really difficult, in short, to discover what those are if you’re not forced to, by capital constraints
Ross:
Of all the things that you’re running in-house, clinical trial operations—I mean, are you contracted out to CROs there? Or are you in direct contact with PIs at sites?
Brian Finrow:
It’s a hybrid. So for our late-stage trial, we’re working with an excellent group called Medpace. They’re based in the Midwest, and I think that’s maybe not a coincidence. They have good Midwestern values, and Seattle has a similar cultural vibe.
Ross:
You come to them with these ideas of efficiency, economy, parsimony—
Brian Finrow:
—and rigor, because that’s a late-stage trial, and they have a lot of recent experience running very similar types of trials in C. diff, and are in direct relationships with a lot of the investigators at hospitals across the country where this trial is happening. So that’s an example where we’re really heavily leaning on the ecosystem.
But for our earlier stage trials, we tend to be much more directly involved. We run them almost exclusively in Australia, which is a bit of a distance and a complexity, but it’s a much more efficient regulatory environment for spinning up early-stage trials and small trials.
And what we really like there—there’s a second aspect of what we like about Australia—is it’s very easy to have kind of a direct relationship with the site PIs. And that’s really important, we think, for early drug development—because if there’s one predominant story about great drugs, it’s that many of them come out of serendipitous observations. Viagra and Rogaine are two famous examples, but there’s many more of these.
Ross:
If it was just by the numbers, endpoints, adverse events, you’d see a lot of adverse events and walk away from it.
Brian Finrow:
So the approach, particularly in the US where there’s so much money available—it kind of complexifies things in a way where it’s difficult to actually see what’s going on directly.
And it’s a real shame, because I suspect we’re missing out on some of these opportunities for serendipitous observation when everything is stripped down and standardized and stamped out, sent out in the standard fashion to the same CROs that run all of the same studies, and they’re all run in the same way. Nobody’s really got enough of the pieces in front of their eyes to see the big picture like this, which is what you need if you’re going to get those serendipitous observations.
“Everyone just has too much money” (1:17:44)
Ross:
This is a question that I’m sure we’ll ask a bunch on the show, but: what is the “original sin” of why clinical trials cost so much and take so long, and we’re spending—how many tens of billions of dollars?—and getting how many drugs out the end?
It sounds like your answer here is that everyone just has too much money. (laughs)
Brian Finrow:
(laughs) I mean, it’s a bit perverse to say that, right? As you know, right now there’s this terrible capital drought happening in biotech.
Ross:
Yes. For a couple of years now, actually.
Brian Finrow:
There’s a lot of companies failing. Last I looked up the numbers, it was something like a quarter or a third of NASDAQ biotech companies have market caps lower than the amount of cash they have in the bank, which is a brutal number. So yeah, it’s a problem.
[Ross: Time for a confession. For the life of me, I don’t understand how you can run a company that trades for less than the money it has in the bank—net of all your obligations and debts—and not face the strong presumption that the right thing to do, as a fiduciary to your minority shareholders, is either to buy them out, or to liquidate the company and give them the cash that the market clearly doesn’t trust you not to waste. I don’t get it! Never have. It’s clear that that’s not how the law works in Delaware, and I’ve made my peace with that, but I still don’t understand it.]
I think, more fundamentally—you’re probably familiar with the term that Jack Scannell came up with, “Eroom’s Law”—Moore’s Law spelled backwards. It’s an observation that—unlike in microchips, which get cheaper and cheaper exponentially—clinical trials and drug development costs seem to get more and more expensive exponentially.
[Ross: Brian, at the point that we taped this, had not seen our first episode with Meri Beckwith where we discussed the very same thing. He just brought it up independently.]
And then Deloitte—the big accounting firm—they run an annual study and crunch the numbers from public biopharma companies, and they have found a pretty persistent secular trend, maybe 20 or 25 years now, of declining profitability. They’re looking at not only the cost of development, but the revenue side, and that’s because the cost to develop one new drug has been going up and the market size achievable with each new drug, on average, has been going down. That’s not a sustainable pattern.
There’s a lot of discourse about what the ultimate drivers are. The one you said—which I think might be part of it—is not typically cited.
Ross:
It’s not typically thrown around, that the problem is everyone having too much money.
Brian Finrow:
But I like your thinking there; let’s imagine that it’s true. What would we imagine happening?
Well, all of those trends hit some kind of asymptote when Covid happened, because not only did everything become more expensive with inflation, but particularly the inputs to biomedical research—even pipette tips. Remember there was this great pipette tips crisis of 2021?
Ross:
And then, when the vaccines were coming out it was—
Brian Finrow:
—the syringes and the glass; there were shortages of everything, and the prices of everything skyrocketed, along with the…the price of lumber and other stuff.
So now we’re very much at the other end—there’s a glut of lab space—
Ross:
—there’s too many pipette tips!
Brian Finrow:
There’s too many pipette tips. (laughs) Basically if you think about Econ 101—what are the factors of production? The layoffs mean there’s a tremendous amount of human capital around. All of the company shutdowns mean there’s a surplus of lab space—in Seattle, there’s a rate of over 40% unoccupied lab space, which is crazy.
What are the other factors of production—? Capital has gotten more expensive, but that’s Jerome Powell’s fault. (laughs) But everything else has gotten cheaper.
Extinction events…and “little mammals scurrying around” (1:21:07)
Brian Finrow:
I think it will be interesting to see—and it’s tempting to draw an analogy to another industry sector that had a similar thing happen to it, where the cost of developing a dot-com in the late ’90s went through the roof. At the time, most of that venture capital money were these huge rounds and immediate IPOs. What was all that money going to? It was going into the hands of Cisco Systems and Microsoft to buy the server chips and software. It was all going to “picks and shovels” kind of places.
Then what happened in 2001, 2002—it was an extinction-level event for not only the dot-coms, but also the VCs that invested in those dot-coms. Most of them didn’t survive. They’re gone, forgotten now.
But what happened afterward, you had little mammals scurrying around, and one of the mammals was Amazon Web Services, wasn’t it? And that set the stage because now, all of a sudden, people didn’t have the capital to buy all the server racks from Microsoft and Cisco—and instead, if you wanted to start an AirBnB or a Netflix, you had no choice. And importantly, you didn’t have the investor community barking down people’s throats to buy Herman Miller Aeron chairs—remember this whole thing? “Spend the money; it’s a gold rush; you’re not spending your money fast enough!”
Ross:
—“you’re not hiring enough people; just hire more people!”
Brian Finrow:
And all of a sudden it became “capital efficiency” and “minimum viable product”, and this whole new way of thinking about it, and there was this whole blossoming. All of the web companies that are dominant today—maybe Amazon is a partial exception, but even them—without AWS, what would Amazon be today? Google, AirBnB, Uber, then the rise of mobile later…
So maybe you’re onto something, and maybe the capital drought in our world will have the same impact in the long run. And boy, wouldn’t that be nice? Because the potential for human flourishing is much greater in biopharma than on streaming media and taxi rides.
Ross:
Maybe less facetiously, there’s Byrne Hobart’s book, Boom, about investment and innovation. He puts forward the hypothesis that you really see a flourishing of new economies, of new realms of activity, when you have this period of overinvestment—you build all of the stuff—then it collapses, and then people have to build up within this ecosystem where there is lab space everywhere, but people have to be lean [in capital]. There is something appealing about that, maybe—we have built all these labs, and maybe something good will happen from all these little mammals scurrying around.
Brian Finrow:
I hope so. I suspect it’s not going to be the “$200 million seed round” company!
Ross:
Some people still can do this [in this environment]; they can raise a billion-dollar fund. The top of this market seems not to have stopped, and maybe it’ll just keep going—but the middle is having a…different…time than they were in 2021.
Wild speculations about the future (1:24:28)
Ross:
I want to end on a few questions about the future—wild speculation allowed, encouraged, and authorized.
If your ability to manufacture cheaply—at scale—scales up to solving enormous tranches of health problems of humanity, what is the scale of that manufacturing enterprise? What does growing that much medical-grade spirulina look like? Is the constraint the raw materials—carbon, nitrogen, oxygen? Is it the doubling time of biology? Is it the photons that you need to pump in for photosynthesis?
Brian Finrow:
It’s the photons.
Ross:
So are you going to need—if this keeps growing, and you serve the world’s metabolic disorder needs, their GI needs, you do a bunch of things for cancer, for respiratory disease—when you’re expanding and you need facility after facility, are you going to need a nuclear reactor or three as the data center folks are learning?
Do you have a vision of the world where this sort of scaled agricultural manufacturing is huge? What is different about that world?
Brian Finrow:
There’s something interesting about our industry. It’s easy to be impressed with these big pharma companies; and some of them do really impressive things—what Novo has done with GLP-1 drugs is pretty remarkable.
And yet, how many trillion-dollar biopharma companies are there?
Ross:
(holds up a “zero”)
Brian Finrow:
Zero, yeah.
And the stability among the incumbents is really pretty profound, too. Who are the hundred-billion-dollar newcomers?
Ross:
Moderna.
Brian Finrow:
Nope, no longer.
Ross:
Well…sure.
Brian Finrow:
Amgen. And Regeneron—they’re 60 billion; we’ll round up.
Here’s another interesting thing, though: who buys Amgen’s products versus who buys Microsoft’s products? And which is a trillion-dollar company? Everybody uses Microsoft products, not just Americans—but it’s Americans, predominantly, who buy Amgen’s products.
[Ross: Indeed, the US is—in round numbers—half the global market for drugs, despite being like 4% of the world’s population.]
So, adding it all up, why can’t we be the first trillion-dollar pharma company? If we’re right and this is as impactful as mammalian cell engineering and the first wave of biotechnology, I think there’s easily enough unmet medical need to support that kind of value creation. But we’re not going to be able to do that unless we’re actually making products that serve the entire world.
The good news here—to your point—is that our products don’t cost as much to make as Amgen’s, and that means we can sell them at a price that the global middle class can afford. It means they have to be great products, because budgets are much tighter there than in the US—but that’s the scale that we think on.
Ross:
Whereas, the global middle class is often an afterthought in medical development—“we’ll get to them after we’ve finished making our money back on Americans.”
Brian Finrow:
The middle class in China is as big as the middle class in the US, it’s just a lower purchasing power parity—
Ross:
—and three times as large.
Brian Finrow:
Yeah, so that’s where we want to go.
How do we service all that? It’ll be a network of manufacturing sites, like what Lilly is doing now—they’re putting in $26 billion. The good news is that—remember—our capex is a small fraction of what theirs is, so it’s definitely doable.
And in the medium term, we can service all of that here, from Washington State. The main input, the main constraint is the photons, we get the photons by conversion of electrons from the hydroelectric plants on the Columbia River that were overbuilt in the ’30s and ’40s and have a lot of surplus power. So this would be great for our state as well; those would be great jobs.
Ross:
You’ll be turning the energy of the Columbia river into medicine for a while yet?
Brian Finrow:
Though, for the future, we really want to have regionalized manufacturing. Of course, we don’t want our photons coming from coal power plants. Not a good look, and anyway, solar is cheaper.
So where do they have cheap power that’s not from an embarrassing source? Because we’re building a consumer brand here, too… Well, Iceland has geothermal. Spain is very nice for solar. The Middle East has a tremendous amount—
Ross:
—of solar, though that’s not usually where we think of the energy as coming from.
Brian Finrow:
Well, a lot of the energy comes from [oil], but solar is becoming increasingly important over there. These are the kinds of places we’re looking to expand in the future.
It’s a little bit cart before the horse—because we don’t have our first approval yet—but that’s all very straightforward from a capital expenditures perspective.
Ross:
And on the other side, on the consumers you’re reaching, if we live in a world where therapeutic proteins—whatever you want—cost $10 a gram, what is the single thing you’re most excited about, that being possible? For human flourishing, for health, for you and your kids…
Brian Finrow:
Our costs at scale are already well below $10 per gram equivalent pure protein. [Ross: lol, oops.]
And at those levels, here’s an idea—I find it discouraging to think about the reimbursement system, how drugs are purchased, distributed, allocated—the policy choices about the allocation made by insurance company formulary managers—
Ross:
—in smoky back rooms, away from—
Brian Finrow:
—and by government agencies, also without a ton of scrutiny. Not just in the US, also in Europe and other places.
So I like thinking about the opportunities for some of our products. Our weight loss product is a great example because it’s already a hybrid market in the US—
Ross:
—as that market is going direct-to-consumer, as well as through the old [insurance] system.
Brian Finrow:
Up until now, it’s been so expensive to make biologic drugs that the only people who can afford them are the ones with rich governments. It’d be nice, if we can get the cost out of it, to sell those at a larger scale—still do a good job for our shareholders who took a lot of risk backing us—but sell it at a price that’s affordable, independent of government solvency, and thereby unlock a much larger market.
That’s an exciting vision for the future. I don’t think many companies are working on it, but I think people are heading in this direction right now. You may have heard about Lilly Direct—they’re selling their GLP-1 drugs directly over the internet, for 30% less. Novo is now getting into the game too, at even more of a discount. So maybe they’re leading the way.
Ross:
Brian Finrow, thank you very much.
Brian Finrow:
Thanks for having me. This was really great, Ross.
